研究概要
当研究チームでは、マウスや酵母などのモデル生物やin vitroの系を用いて、神経科学、構造生物学、遺伝学、プロテオミクスなどの様々な手法から、タンパク質の構造異常が引き起こす神経変性・精神疾患の病態解明、プリオンの形成・伝播における分子構造基盤の解明、さらに、アミロイドなどのタンパク質凝集体の新たな生理機能の発見を目指した研究を展開しています。
プレスリリース
| 2024.3.12 | 
|
| 中川幸姫研究員が2023年度理研・桜舞賞(研究奨励賞)を受賞しました! |
|---|
| 2024.1.22 | 
|
| 私たちの研究が「CBS Magazine Fall and Winter 2023 Vol.6」で紹介されました! |
|---|
| 2023.12.21 |
| 玉井真悟さん(院生)が第46回日本分子生物学会のサイエンスピッチで優秀発表賞を受賞しました! | 2023.11.28 |
|---|
| 佐藤海研究員がCBSリトリートで口頭発表者に選ばれ、研究発表を行いました! |
| 2023.11.10 |
| 中川幸姫研究員と玉井真悟さん(院生)がAPPS2023国際会議でともにベストポスター賞を受賞しました! |
|---|
| 2023.11.9 |
| 野村高志研究員がAPPS2023国際会議で口頭発表しました! |
|---|
| 2023.9.1 | |
| 私たちの研究が国際広報誌「RIKEN Research 2023」のResearch Highlightで紹介されました! ~An over-reaction when protein synthesis stalls can lead to neurodevelopmental disorders in mice~ |
|---|
| 2023.5.24 | 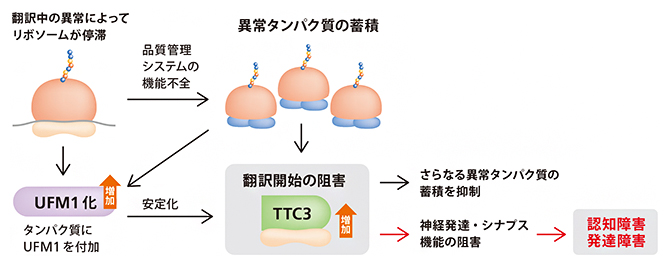 |
| 私たちの研究が「クローズアップ科学道」で紹介されました! ~神経疾患の鍵を握る細胞の“品質管理”~ |
|---|
| 2023.5.19 |  |
| 玉井真悟さんがThe 27th Biophysics Conference (Hualien, Taiwan)で銅賞を受賞しました! |
|---|
| 2023.03.14 | 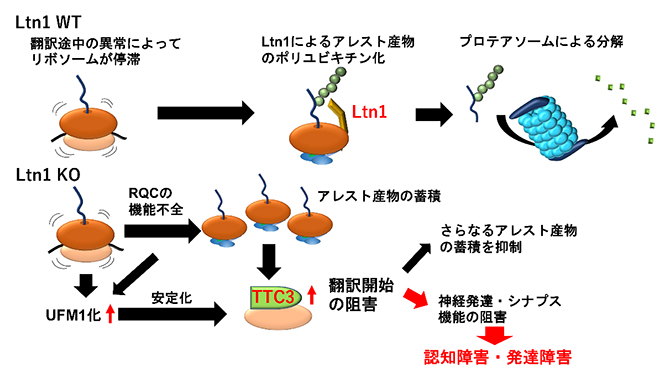 |
| リボソーム品質管理の破綻と神経疾患
-TTC3タンパク質の異常な蓄積による認知・発達障害の発現- |
|---|
| 2022.12.16 |
| 小見悠介研究員がAPPS2022国際会議でポスター賞(第二位)を受賞しました! 佐野俊春研究員がAPPS2022国際会議でショートプレゼンテーション賞を受賞しました! |
|---|
| 2022.11.29 | ||
| 玉井真悟さんがクロススケール新生物学若手の会で最優秀発表賞を受賞しました! 野村高志研究員がクロススケール新生物学若手の会で優秀発表賞を受賞しました! |
|---|
| 2022.10.02 |  |
| Nayan SuryawanshiさんがUCSF-CBS Young Investigator Exchange Travel Awardに採択され、UCSFのリトリート(Pacific Grove, CA)で研究発表を行いました! |
|---|
| 2022.09.20 |
| 持田啓佑研究員がJSTさきがけ研究者([高次構造体]細胞の動的高次構造体)に採用されました! |
|---|
| 2022.02.18 | 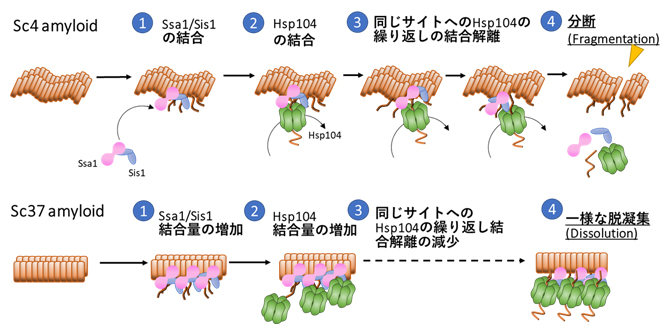 |
|
| アミロイドの脱凝集メカニズムを解明 −アミロイド構造に依存した脱凝集機構が明らかに− | News & Views Picturing protein disaggregation |
理研リサーチニュース (2022.5.26) Three chaperones coordinate the breakup of amyloid fibrils in yeast |
|---|
| 2021.10.1 |
| 私たちの研究課題がAMED-CRESTの研究開発領域「プロテオスタシスの理解と革新的医療の創出」に採択され、研究開発代表者として参画します。 |
|---|
| 2021.09.10 |
| 学術変革領域(A)「クロススケール新生物学」(代表:東大・医 吉川雅英教授)が採択され、私たちの研究室が計画班員として参画します。 |
|---|
| 2020.04.14 | |
| プリオン感染における「種の壁」を解明 -短い天然変性領域の揺らぎによる感染制御機構が明らかに- |
理研リサーチニュース (2020.7.17) A short segment of a prion protein plays a critical role in its susceptibility to cross-species prion transmission |
|---|
| 2019.04.26 | |
| 細胞分裂時のタンパク質分配の偏りを網羅的に解析 -セプチンによる細胞内区画化機構などを解明- |
|---|
| 2019.04.11 | |
| オートファジー機能の欠損が自閉症様行動を誘導 -発達障害や精神疾患の克服に向けた新たな治療戦略に貢献- |
|---|
 |
 |
 |
 |
 |
|
 |
 |